

Welcome to the 9th edition of Gradient Ascent. I’m Albert Azout, a prior entrepreneur and current Partner at Cota Capital. On a regular basis I encounter interesting scientific research, startups tackling important and difficult problems, and technologies that wow me. I am curious and passionate about machine learning, advanced computing, distributed systems, and dev/data/ml-ops. In this newsletter, I aim to share what I see, what it means, and why it’s important. I hope you enjoy my ramblings!
Is there a founder I should meet?
Send me a note at albert@cotacapital.com
Want to connect?
Find me on LinkedIn, Angelist, Twitter
There is something quite magical about the application of computational methods to complex biological phenomena. Recently, there has been an acceleration of deep learning and statistical learning approaches aimed at several important problem areas in the biological realm.
One such problem area is the elusive protein folding problem.
DNA structure is well-understood (discovered by Watson and Crick in the early 1950s). The same, however, cannot be said about protein structure. While one of DNA’s primary purposes is to encode proteins—nanomachines that physically carry out most of our biological functions—progress towards predicting protein structure has been a long and incremental journey.
In general, the protein folding problem aims to answer three questions:
What is the physical folding code of proteins? How is 3D structure determined from the physiochemical properties encoded in the 1D structure of amino acid residues?
What is the folding mechanism? A polypeptide chain (chain of amino acids) has a huge combinatorial set of conformations (folding poses), so how do proteins fold so fast to lock into their native structure?
Can we use computational methods to predict protein structure? If so, such methods would circumvent experimental protein structure determination (i.e. x-ray crystallography, NMR structure determination, etc).
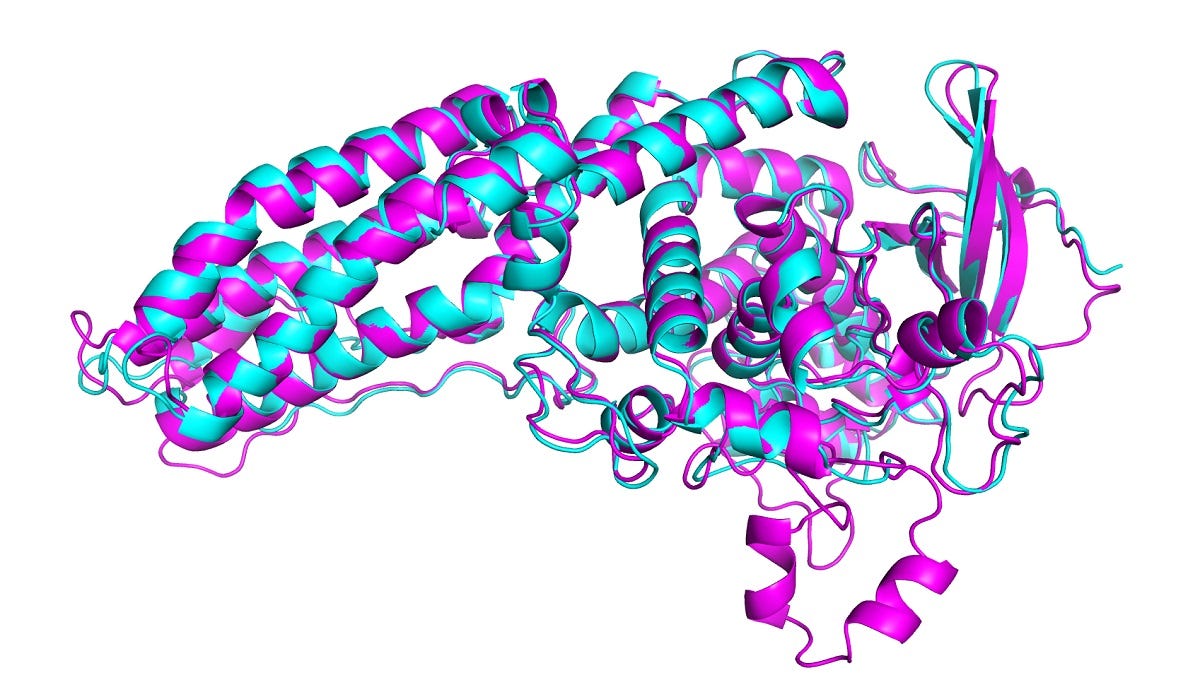
Historically, computationally, molecular dynamics simulation (MD) techniques have been employed in conjunction with experimental structure determination. In MD, a molecule is described as a series of points (atoms) connected by springs (bonds). To describe the time evolution of bond lengths, bond angles and torsions (and also the non-bonding van der Waals and elecrostatic interactions between atoms) one uses a force field (FF)—a collection of equations and associated constants designed to reproduce molecular geometry and tested structures. FF models explore the ways in which proteins progress through conformational states towards lower free energies (thermodynamically stable states, see below, experimental structures are in red, computed structures in blue, with the protein name, PDB identifier, and error between the predicted and experimental structures).
To capture a single folding event, long MD simulations are required, which drive extreme computational cost and/or the need for a specialized hardware accelerators. Enhanced sampling methods are typically utilized to accelerate the dynamics of such systems in simulations (this is one application of quantum computing).
MD approaches are strictly physical.
Now enters deep learning…
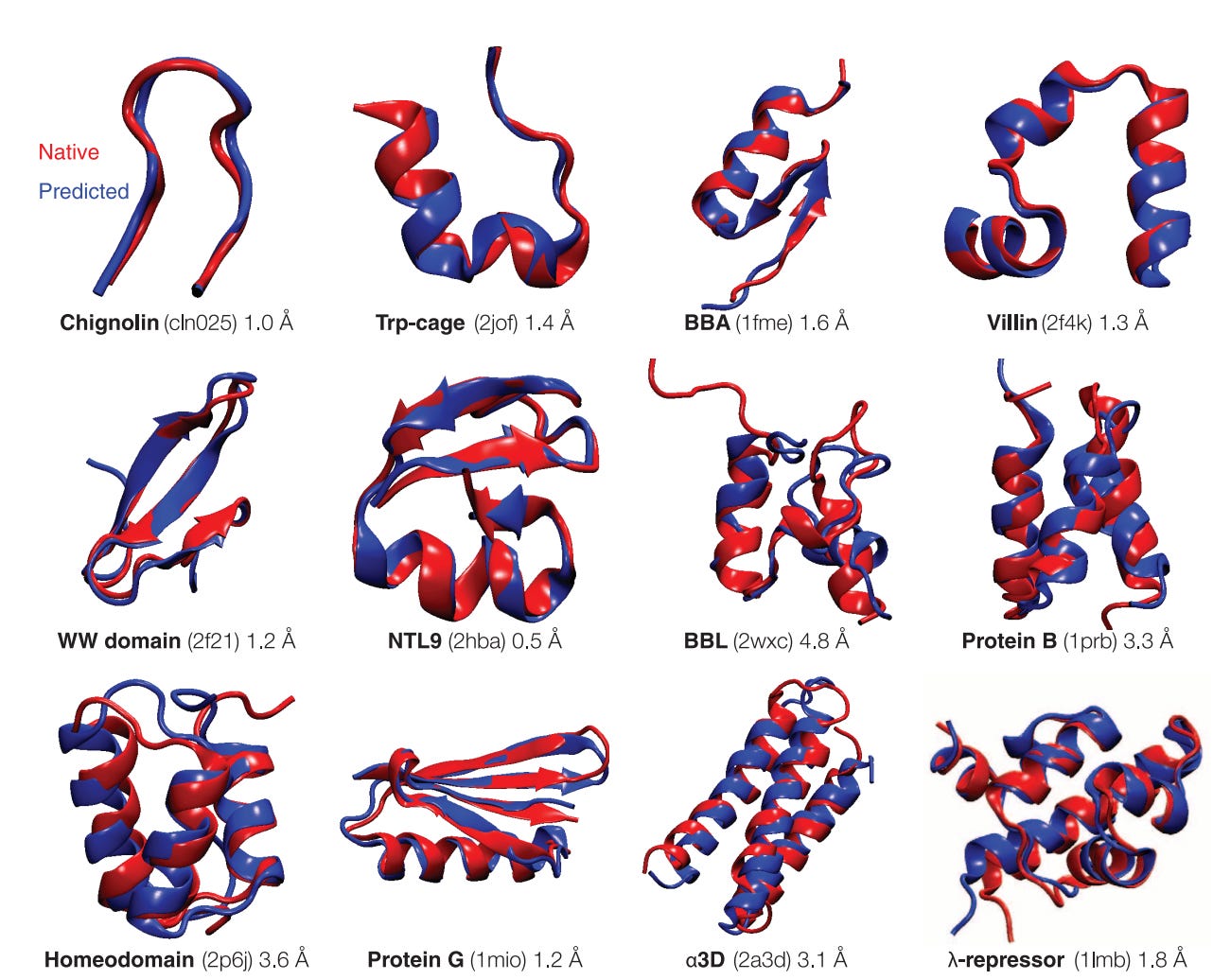
Deep learning provides a shortcut: instead of simulating physics or performing statistical optimizations (sampling) on a simulated system, the system can directly recognize sequences that indicate particular structural conformations. In 2018, DeepMind’s AlphaFold significantly beat the best competitors in the CASP13 competition using such an approach.
This week a new breakthrough was announced from DeepMind (‘The game has changed.’ AI triumphs at solving protein structures”). In the recent CASP14 competition, AlphaFold was evolved to fully model proteins as spatial graphs (amino acid residues are the nodes and edges are connections between residues).

“For the latest version of AlphaFold, used at CASP14, we created an attention-based neural network system, trained end-to-end, that attempts to interpret the structure of this graph, while reasoning over the implicit graph that it’s building. It uses evolutionarily related sequences, multiple sequence alignment (MSA), and a representation of amino acid residue pairs to refine this graph (source).”
The paper is yet to be released but the new AlphaFold provides an incredible amount of prediction accuracy.
A few things are immediately fascinating to me:
The complexity of the network topology, including various informational modules that learn and reason from each other (all likely trained jointly).
The reasoning about proteins as spatial graphs—explicitly predicting structure from sequence versus needing to simulate dynamics.
The hope is that this new model will help the world devise drugs that bind to proteins as well as the ability to synthesize proteins that have desired properties, thereby speeding the development of enzymes (make biofuels, degrade waste plastic, etc).
Exciting times in life sciences! 🧬
Quasi Sequiturs
A really amazing and must-read book written by James Watson, the co-discoverer of the structure of DNA…

Disclosures
While the author of this publication is a Partner with Cota Capital Management, LLC (“Cota Capital”), the views expressed are those of the writer author alone, and do not necessarily reflect the views of Cota Capital or any of its affiliates. Certain information presented herein has been provided by, or obtained from, third party sources. The author strives to be accurate, but neither the author nor Cota Capital do not guarantees the accuracy or completeness of any information.
You should not construe any of the information in this publication as investment advice. Cota Capital and the author are not acting as investment advisers or otherwise making any recommendation to invest in any security. Under no circumstances should this publication be construed as an offer soliciting the purchase or sale of any security or interest in any pooled investment vehicle managed by Cota Capital. This publication is not directed to any investors or potential investors, and does not constitute an offer to sell — or a solicitation of an offer to buy — any securities, and may not be used or relied upon in evaluating the merits of any investment.
The publication may include forward-looking information or predictions about future events, such as technological trends. Such statements are not guarantees of future results and are subject to certain risks, uncertainties and assumptions that are difficult to predict. The information herein will become stale over time. Cota Capital and the author are not obligated to revise or update any statements herein for any reason or to notify you of any such change, revision or update.